
30 pages, 8.2 MB

This briefing’s findings are summarized in the Executive Summary, COVID in California How Government Regulations Created Critical Healthcare Shortages—Permanent Patient-Centered Reforms Needed.
Contents
- Overview
- I. The Pathologies of Government: Harming Patients by Granting Regulatory Favors to Healthcare Interest Groups
- II. Shortages of Personal Protective Equipment and Medical Equipment
- III. Shortages of Healthcare Workers
- IV. Botched COVID-19 Testing
- V. Vaccine and Therapeutic Antiviral Drug Development
- VI. Patient-Centered Recommendations
- VII. Conclusion: The Moral Imperative Not to Return to the Bad Old Days
Overview
On January 20, 2020, the first case of Coronavirus Disease 2019 (COVID-19) was confirmed in the United States in the state of Washington. A thirty-five-year-old man returned to Washington after visiting family in Wuhan, China, and later was diagnosed with COVID-19. The new infectious disease, caused by a novel coronavirus called Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), can drastically impair the functioning of an individual’s respiratory system.
The first non–travel-related (“community spread”) case of COVID-19 in the United States was in California—confirmed on February 26 in Solano County. A resident had become sick on February 13 despite having not traveled to any areas with known infections. After the contagion spread, California governor Gavin Newsom (D) signed Executive Order N-33-20 on March 19, 2020, a statewide shelter-in-place order resulting in months of quarantine for tens of millions of Californians. Eight days earlier, on March 11, 2020, the World Health Organization (WHO) declared the COVID-19 outbreak a pandemic.
During the initial months of the outbreak, it quickly became apparent that California’s state government, along with the federal government, was woefully unprepared, despite many past warnings. A 2005 report published by the US Homeland Security Council warned of the potential threat of a widespread viral pandemic: “States and communities should have credible pandemic preparedness plans to respond to an outbreak within their jurisdictions. The private sector should play an integral role in preparedness before a pandemic begins and should be part of the national response.”
Unfortunately, officials at all levels of government failed to heed the call to action. Perhaps even worse, regulatory barriers prevented rapid and efficient private-sector responses, which fueled the crisis and harmed healthcare consumers. Government-imposed regulatory straitjackets created breaking points in California’s healthcare system and are the focus of this Golden Fleece report.
Governments failed to plan effectively, failed to prepare, and failed to respond quickly or at all. When governments did respond, they found themselves playing a high-stakes game of catch-up, as many regulations imposed for years, even decades, prevented quick, efficient, and flexible responses in four key areas: (1) acquiring sufficient quantities of medical supplies and equipment, (2) expanding the healthcare workforce across occupations, (3) developing and distributing a sufficient number of tests, particularly rapid nonprescription at-home tests, and (4) developing and distributing effective vaccines and drug therapeutics.
State and federal governments belatedly removed regulatory barriers that prevented rapid responses to the outbreak. Governor Newsom said in March 2020 that the emergency actions “will increase California’s healthcare capacity and help facilities treat more patients.” He implied that government policies knowingly have reduced California’s healthcare capacity and harmed the ability of facilities to treat more patients. As shown in this report, an extraordinary number of government regulations and regulatory agencies have impaired healthcare services.
The report spotlights the lessons that must be learned from the COVID-19 outbreak, lest we repeat past mistakes. It explores why harmful regulations existed in the first place, who benefited from them, who was harmed, and the gains from removing the regulations, especially the benefits to healthcare consumers. It is important to keep in mind that if removing the harmful regulations during a crisis is the moral action to help patients and save lives, it would be immoral to reimpose the same regulations when the crisis ends. The emergency liberalizations have helped today’s healthcare consumers, and they would help consumers in the future too, if made permanent. Consumers are better off without the regulatory barriers—safety and service would be enhanced.
For enacting or enforcing regulations that created critical healthcare shortages and prevented a fast and efficient response by public and private entities to COVID-19, the Independent Institute awards its eleventh California Golden Fleece® Award to federal and state officials and regulatory agencies, including the California legislature; Governor Newsom; past California governors; US Centers for Disease Control and Prevention (CDC); the US Food and Drug Administration (FDA); US Centers for Medicare and Medicaid Services (CMS); US Customs and Border Protection (CBP); labor unions, professional associations, and boards representing medical personnel; and the nation’s flawed intellectual property laws.
California would be in a better position today to handle the surge in COVID-19 patients if governments had not maintained for decades artificial restrictions on medical supplies, the healthcare labor force, and test and drug development. Rather than simply waiving those regulations temporarily during the pandemic, California lawmakers and federal officials ought to abolish them permanently because the regulations harm healthcare consumers, especially low-income Californians and those in poor health. Californians deserve a nimbler, more efficient, and more effective healthcare system at all times, not just during a global pandemic. If it works during the present crisis, it will work when the crisis is over.
I. The Pathologies of Government: Harming Patients by Granting Regulatory Favors to Healthcare Interest Groups
The healthcare industry is among the most regulated sectors of the US economy, with an alphabet soup of federal, state, and local government agencies that regulate various aspects of the industry, imposing mandates, fees, and fines. In addition, every healthcare occupation and type of provider—for example, hospitals—have its own professional association (national and/or state) that lobbies for the narrow interests of their members.
The conventional argument for the labyrinth of healthcare rules and regulations is to protect the health and safety of consumers and workers in the industry. That argument contains a grain of truth—everyone wants to be safe from dangerous drugs, devices, and doctors. But a large body of evidence in the economics and political science scholarly literature concludes that the true motivation for much regulation is to protect incumbents from outside competition.
Popularized by Nobel laureate economist George Stigler (“The Theory of Economic Regulation”) and economist Sam Peltzman (“Toward a More General Theory of Regulation”), the theory of economic regulation argues that interest groups compete within a political context to influence government regulation in their favor. Often, rivalry among interest groups results in producer groups effectively “capturing” regulatory agencies and using government force to prevent competition, benefiting themselves at the expense of consumers.
In the healthcare sector, interest groups represent occupations (e.g., physicians, nurses, pharmacists), providers (e.g., hospitals, clinics, urgent care centers, pharmacies), and suppliers (e.g., medical device manufacturers and drug developers). Each interest group typically has its own lobbying entity. Examples include the California Hospital Association, California Medical Association (physicians), California Pharmacists Association, California Registered Nurses Association, and many others. Each fights for the interests of its members. Unorganized consumers are left to fight for themselves.
By supporting government regulations that raise artificial barriers to entry and restrict competition, interest groups are able to increase their incomes, profits, and political influence. They have learned over time that the surest means of blocking competition is to “capture” the governmental regulatory apparatus and use it to create protected cartels. The cartels benefit the narrow self-interests of privileged groups but harm consumers and innovators.
If you think the primary purpose of healthcare regulations is to ensure patients’ safety, ask yourself, as you read this report, the following: Why were safe and effective drugs, medical devices, and medical equipment that were approved and used elsewhere excluded from California? Why were skilled healthcare professionals with years of experience who are licensed to work elsewhere banned from working in California? Why were certain point-of-care resource allocations, such as redesignating beds or reassigning nurses, that would improve patient care outlawed in California? The answer is that the regulations have little to do with safety and everything to do with protecting incumbents from competition—in fact, the regulations have reduced patient care in many ways, as highlighted in this Golden Fleece report.
Significant costs are imposed on others when healthcare interest groups use regulatory agencies to benefit themselves. In addition to the enormous enforcement and administration costs, innovative lifesaving drugs and devices are delayed. Competition among providers is short-circuited, thus increasing prices, and reducing access to higher-quality care.
Consumers are harmed by interest groups capturing the regulatory machinery of governments, and COVID-19 exposed the ways that regulations are used in California to benefit narrow interest groups at the expense of consumers at large. COVID-19 taught a crash course to Californians on the downside of excessive regulation and the benefits of liberalization.
II. Shortages of Personal Protective Equipment and Medical Equipment
Personal protective equipment (PPE) was in short supply during the first wave of COVID-19, and it continued to be in short supply throughout much of 2020 until new supply chains were established. PPE includes hand sanitizers, masks, shields, goggles, respirators, gowns, and gloves. Critical shortages of other medical supplies and equipment, such as swabs, reagents, and ventilators, also became evident.
Owing to regulatory barriers, importing medical supplies (both PPE and equipment) from abroad always has been a challenge. And getting new US manufacturers certified to produce medical supplies has also been slow and inefficient, with miles of red tape and huge bureaucratic obstacles. The pandemic exposed the deadly consequences of the regulatory hurdles and the need to ease government restrictions permanently in order to speed responses and save lives.
Many long-exisiting regulations act as nontariff barriers to foreign-made equipment or as protections for incumbent US manufacturers from other domestic suppliers. The regulations include licenses and certifications, quality control checks of equipment during manufacturing and transit, labeling requirements, and barriers to entry into American markets, even when, in the midst of a crisis, the equipment could have been extremely beneficial, even lifesaving.
In its review of the 2003 SARS outbreak, the US General Accounting Office reported that “most hospitals lack the capacity to respond to large-scale infectious disease outbreaks. ... [F]ew hospitals have adequate medical equipment, such as the ventilators that are often needed for respiratory infections such as SARS, to handle the large increases in the number of patients that may result.”
In 2009, years before the nationwide shortage of N95 masks in the first months of the novel coronavirus outbreak, the US Occupational Safety and Health Administration sounded the alarm: “It is expected that there will be a worldwide shortage of respirators if and when a pandemic occurs. Employers and employees should not count on obtaining any additional protective equipment not already purchased and stockpiled. Therefore, it is important for healthcare facilities to consider respiratory protection for essential personnel to assure that employees are ready, willing, and able to care for the general population.”
Over the course of the H1N1 swine flu outbreak of 2011, which was smaller than COVID-19 in terms of attributable deaths, 100 million masks were drawn from the Strategic National Stockpile but never replenished.
At the state level, despite multiple warnings over many years, the state of California allowed its emergency stockpile of twenty-one million N95 masks to expire. So dire was the situation that in a March 3, 2020, California Department of Public Health (CDPH) press release, the CDPH resorted to directing attention to the CDC’s guidelines on how to reuse N95 respirators.
The depletion of stockpiles led to critically short supplies in several California communities that became virus hot spots, such as the San Francisco Bay Area. An outpatient nurse based in Fremont, California, told reporters that she was instructed to reuse her disposable surgical mask every day, despite being repeatedly exposed to patients potentially infected with the coronavirus. Nurses caring for patients during normal times are expected to use a dozen masks per day, with standard protocols dictating the disposal of any mask that might be contaminated.
The cost of failing to stockpile and properly supply healthcare workers with PPE was enormous. A study conducted by researchers at the University of California, Berkeley, concluded that dozens of deaths in California were attributed to inadequate protective equipment for essential frontline workers. Additionally, 15,800 COVID-19 cases among healthcare and other essential workers and their household members were potentially avoidable with proper PPE.
The same study also found that California’s lack of PPE had a significant fiscal impact on the state, and it caused significant delays in the provision of routine medical services. Gov. Newsom’s Executive Order N-33-20 of March 19, 2020, prioritized PPE for potentially vulnerable populations in California hospitals. PPE supplies were diverted away from care unrelated to COVID-19, causing major disruptions, especially in preventative care and certain nonemergency elective surgeries. The state had to ration scarcity rather than manage abundance.
Although the state of California’s preparedness was deficient, it is not the only culprit. Federal government regulations also impeded efficient supply chain operations. Any PPE that is intended for use to prevent illness and is manufactured abroad must go through a strict process of submitting information to the FDA at time of entry into the United States (equipment that is not intended to prevent illness, such as many safety items in industrial workplaces, are not subject to the same regulations). Before the outbreak of COVID-19, the application process for importing PPE was the same as the requirements for importing any medical device; that procedure was tedious, costly, and overseen by US CBP officials.
Importation typically involves an importer and a foreign exporter working with a “customs broker,” a company that assists importers in navigating the bureaucracies of the CBP and FDA, to provide detailed information, such as package size, country of origin, and specific medical uses. Arguably, the most complex step in the process involves the device’s classification on the FDA’s I through III scale, which is “based on the level of control necessary to assure the safety and effectiveness of the ,” and then cooperating with a series of inspections by the CBP. The process results in costly, long delays at points of entry and complex barriers that reduce import competition. Those requirements obviously harmed healthcare workers and healthcare consumers during the COVID outbreak when they needed abundant supplies quickly of adequate medical equipment.
All medical devices in the United States are classified into one of three categories: I, II, or III. The categorical assignment of each device is based on possible risks. Factors that may be used to determine categorization are such things as the length of time the device will be used, and whether or not the device is implantable, used in surgery, or contains medical substances.
Class I devices pose the least risk and include such things as dental floss, tongue depressors, and adhesive bandages. The devices must be registered, but they do not require FDA premarket approval. Class II devices are considered to be more risky than Class I and include such things as contraceptives, biopsy needles, and powered wheelchairs. The FDA requires premarket notification for some Class II devices. Considered to be of highest risk are Class III devices, which require FDA premarket approval. Examples of Class III devices are implantable pacemakers and aortic stents.
Key belated federal government responses
In response to COVID-19, the FDA issued several Emergency Use Authorizations (EUAs) and guidelines that effectively lowered barriers to importing PPE and medical devices; they also eased restrictions on US manufacturers who wanted to supply the critically needed equipment. Using authority granted by federal laws, the FDA created an expedited route using its EUA powers. According to the FDA, “During a public health emergency, the FDA can use its Emergency Use Authorization (EUA) authority to allow the use of unapproved medical products, or unapproved uses of approved medical products, to diagnose, treat, or prevent serious or life-threatening diseases when certain criteria are met, including that there are no adequate, approved, and available alternatives.”
EUAs are intended “to help make medical products available as quickly as possible by allowing unapproved medical products to reach patients in need when there are no adequate, FDA-approved and available alternatives” (visit here for a list of FDA EUAs covering devices, diagnostic tests, PPE, ventilators, and more).
One FDA EUA waives some holding and manufacturing regulations on face masks. Other actions focus on medical devices, sterilizers, disinfectant devices, air purifiers, ventilators, anesthesia gas machines modified for use as ventilators, positive pressure breathing devices modified for use as ventilators, ventilator tubing connectors, and ventilator accessories. For ventilators, some authorizations allow modifications to various components without FDA approval, for example, software updates and battery or mechanical swaps. With their backs against the wall, flexibility and rapid response suddenly became a priority for government officials scrambling to catch up to circumstances.
The EUAs exempt the products from premarket FDA review, unless they are intended for high-risk use. For example, medical device sterilizers are classified as exempt from review, unless they are for sterilizing endoscopes.
How do the FDA’s EUAs expedite importation? If an authorized device or PPE needs to be imported, the importers of equipment authorized under an EUA do not need to register and , which is the process for documenting the device’s registration, proprietary name, product code, and submission type. “” prior to importation, notes the CBP, which fast-tracks the importation of authorized supplies and equipment. (For details, see the FDA’s “.”)
A June 30, 2020, US CBP document provides an extensive summary list of the PPE and medical equipment subject to FDA EUAs, and, therefore, eligible for fast-track importation. It took a pandemic to open markets to greater global sourcing and competition, but not until critical shortages emerged.
In the face of shortages and supply chain breakdowns, President Donald Trump (R) invoked the 1950 Defense Production Act to compel the production of specific products, as was done during World War II under the War Powers Acts of 1941 and 1942. Although the act compromised the autonomies of private companies, it was invoked in this instance to compel General Motors to manufacture ventilators. CBP also was authorized to block exportation of PPE, with the Federal Emergency Management Agency (FEMA) deciding how the intercepted PPE would be allocated. The law also authorized mobilization of the military to combat COVID.
Key belated California government responses
On June 5, 2020, Gov. Newsom signed Executive Order N-68-20 to allow companies that had not been licensed by the CDPH temporarily to meet demands unfilled under existing regulations. Newsom’s office said that the order would “help increase the availability of over-the-counter drugs, such as hand sanitizer, and medical devices, such as respirators, ventilators, and masks, which are in demand due to the COVID-19 pandemic.” Companies still had to receive a designation from the state and self-certify that they were FDA compliant.
California, and other states, also struggled to procure adequate numbers of ventilators, key to treating severely ill COVID-19 patients. Primarily afflicting the respiratory system, a COVID-19 infection means that a ventilator, which moves air in and out of the lungs of a patient who is unable to breathe, is a potentially lifesaving device. Of the patients with COVID-19 in intensive care units, most require ventilators.
California, which had an emergency stockpile of ventilators in response to the 2006 avian flu (H5N1) scare, dismantled its reserve. And officials ignored calls to stockpile ventilators and to help prepare facilities to accommodate more ventilators.
A 2015 study published in the journal Disaster Medicine and Public Health Preparedness found that the “current US healthcare system may have limited capacity to use additional mechanical ventilators during a large-scale public health emergency. Emergency planners need to understand their healthcare systems’ capability to absorb additional resources and expand care.”
In 2017, the CDC warned, “Substantial concern exists that intensive care units (ICUs) might have insufficient resources to treat all persons requiring ventilator support. Prior studies argue that current capacities are insufficient to handle even moderately severe pandemics and that sentinel reporting and model-based decision-making are critical for managing limited resources.”
In the weeks following the first confirmed COVID-19 cases in California, the number of infections was small in comparison to hot spots, both nationally and internationally. Newsom announced that the state would lend five hundred ventilators to the national stockpile, with the state of New York being the primary recipient. After a surge of cases in Michigan a week later, California loaned Michigan fifty ventilators in addition to the five hundred loaned to the national stockpile. Newsom acted after assuring Californians that sufficient reserves of ventilators were available to “meet the needs” of the state. At the same time, however, health officials in Santa Clara County began asking for donations of ventilators—going so far as offering $1,000 for broken ventilators because the county projected a shortage.
In conjunction with the PPE shortage, the ventilator shortfall hit California especially hard. The estimated number of ventilators available to the state’s hospitals was 7,500. Newsom stated in March 2020, that to address the high demand, California would need a total of ten thousand ventilators. By the end of July, California was still far below that goal. To make matters worse, 170 ventilators received directly from the federal stockpile were reported defective. Scrambling, state officials decided to outsource the refurbishment of the broken ventilators to Bloom Energy, a San Jose–based fuel cell company. But as a consequence of California’s procurement of more ventilators, other states were left competing for the same scarce resources.
After it was announced that California would spend nearly $1 billion for 200 million medical masks monthly, state officials from around the country found themselves paying skyrocketing prices in a bidding war with California for masks and ventilators. At one point, the state of New York reported paying fifteen times the normal price to obtain some medical equipment. In a press conference, Andrew Cuomo, the governor of New York, said, “It’s like being on eBay with fifty other states bidding on a ventilator. And then, FEMA gets involved and FEMA starts bidding! And now FEMA is bidding on top of the fifty! So, FEMA is driving up the price. What sense does this make?”
At the end of August 2020, the California legislature passed a bill that allocates $250 million to the CDPH for the purpose of creating a PPE stockpile. The bill stipulates that the state has one year to acquire a ninety-day supply, while healthcare facilities are required to amass their own forty-five-day supplies. But it was too little, too late—widespread harm already had occurred.
Summary
Despite multiple warnings years before the COVID-19 outbreak, governments did not stockpile critical supplies adequately, and perhaps more important, governments maintained regulatory barriers that prevented fast and efficient market responses by private-sector entrepreneurs wanting to alleviate emerging shortages. Governments protected incumbent manufacturers of PPE and medical devices from domestic and foreign competition, using public safety as the justification. Ironically, public safety became the victim. With their backs against the wall, officials adopted emergency liberalizations—but, by then, much harm already had occurred to healthcare patients and workers.
In an effort to protect incumbent US manufacturers of PPE, ventilators, and other medical equipment, the federal government and California state government erected barriers to the rapid importation of foreign-made products and to rapid manufacture by domestic producers. That political strategy proved to be deadly and destructive during the COVID-19 pandemic. While regulations benefited companies such as 3M over the years, they were deadly obstacles to obtaining needed supplies quickly after COVID struck. Public safety was sacrificed to benefit politically connected interest groups.
The liberalizations that eventually were adopted should be continued in perpetuity in order to help healthcare patients and medical personnel—pandemic or no pandemic. If it is the right thing to do now, it is the right thing to do in the future.
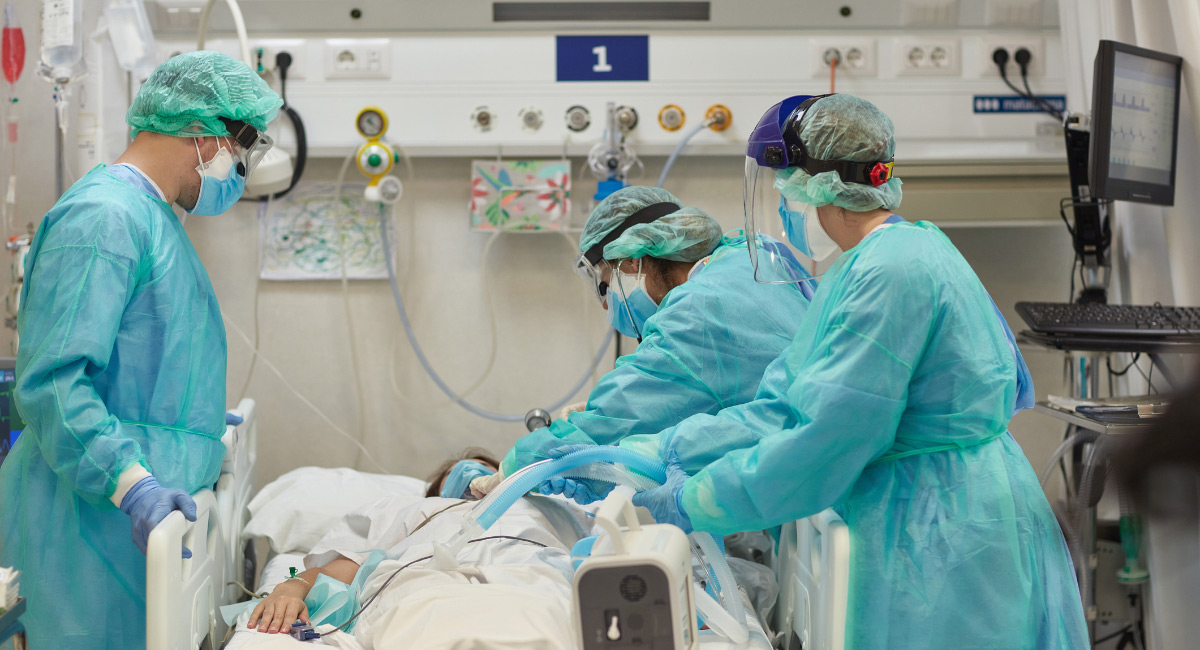
III. Shortages of Healthcare Workers
Regardless of the adequacy of medical supplies, if enough healthcare workers are not available, patient care will suffer. For example, it does not matter how many ventilators a hospital has if the number of respiratory therapists required to properly operate the machines and monitor intubated patients is inadequate. A single ventilator patient usually occupies the attention of a physician, a nurse, and a respiratory therapist.
Additionally, intensive care unit (ICU) beds can be manufactured relatively quickly but are useful only if sufficient critical care nurses and physicians are available. “The issue isn’t only that of facilities—we could set up several—it’s one of staffing,” said Yuba County, California, spokeswoman Rachel Rosenbaum in a December 2020 email to the Sacramento Bee. “Hospitals cannot expand more ICU or regular beds unless they have the nursing staff to take care of those patients, and right now, the hospital is dealing with severe staffing shortages.” Many parts of California experienced critical shortages of ICU doctors and nurses amid the surge of COVID-19 cases from November 2020 into 2021.
It is important to keep in mind that California suffered from severe shortages of healthcare workers before the outbreak. The pandemic exacerbated the shortages, which became worse from early 2020 through early 2021. The shortages largely were avoidable if not for the staggering number of government rules that restrict the supply of qualified healthcare personnel artificially and limit their optimal allocation.
In order to work in the healthcare industry in the United States, physicians, nurses, and others must graduate from approved training programs and obtain various licenses and certifications, which vary from state to state. Governments enforce rules that artificially restrict new entrants into the healthcare professions. Incomes rise for established practitioners, but access to care is reduced and prices for consumers rise.
On top of those restrictions are scope-of-practice rules that dictate what licensed professionals can do once they are in the workforce. Supervisors and administrators at points of care—e.g., hospitals and clinics—decide which employees to hire and which to promote, but daily work arrangements are shaped by scope-of-practice rules, government-mandated staffing requirements, and labor union contracts. The healthcare labor market is a regulatory thicket.
The stated justification for strict government controls is to ensure patient safety, but the controls go beyond what is needed and do not allow market signals to determine where additional workers are required and with what skillsets. Once in place, the regulations do not allow rapid adjustment to changing circumstances. Healthcare workers who are qualified and available to work are excluded from the market; thus, the supply of healthcare workers is artificially restricted, which is especially harmful during a crisis.
The regulations have the effect of limiting the supply of healthcare personnel, creating labor shortages, reducing access to high-quality care at affordable prices, and reducing competition among providers. Healthcare personnel receive higher incomes at the expense of patients. As has become evident during the pandemic, the state’s heavy-handedness undermines rapid mobilization and harms patients because it slows responses to changing circumstances and changing patient needs inside hospitals, clinics, and elsewhere.
As the number of COVID-19 patients escalated rapidly in California, stresses appeared in the healthcare labor force. In anticipation of high rates of hospitalization, in March 2020, Gov. Newsom called for an additional thirty thousand hospital beds, a marked increase from a previous estimate of twenty thousand more beds. More patients filling more beds also means that more workers must be trained, certified, recruited, and hired, which takes time.
Scott Casanover, senior vice president of governmental affairs at West Coast University and American Career College, which operate nursing schools, lamented in March, “We started appealing to the [regulatory] board three weeks ago saying this will be a huge problem if we can’t graduate nurses.” As an emergency room nurse told KTVU Fox 2 in the San Francisco Bay Area, “We’re always short of nurses. We’re doing the best we can in the wake of COVID-19.” Adequate levels of nursing staff, particularly those with expertise in respiratory illnesses, are critical to containing and treating an infectious disease such as COVID-19.
Long before California’s statewide shelter-in-place order, alarm bells were ringing about labor shortages in healthcare. A 2001 report by the Center for the Health Professions at the University of California, San Francisco (UCSF), projected healthcare labor shortages in 2020. A particular area of concern was nurses. The authors belabored the point that “without nursing, many of the services that are taken for granted would not be available or would be seriously compromised.” A prophetic warning for California.
Following up in a more recent report, Joanne Spetz with the Healthforce Center at UCSF pointed out in 2018 that regional shortages already were evident throughout California: “The Fall 2017 Survey of Nurse Employers found that many Chief Nursing Officers are experiencing difficulty recruiting RNs [registered nurses] for specialized positions and that more than 85 percent of hospitals reported demand for RNs being greater than the available supply (Chu, Bates, and Spetz 2018). ... There is variation across regions in the reported difficulty of finding qualified staff, with some employers suggesting there is a surplus of recently graduated nurses and others indicating severe shortfalls of nurses at all levels of experience.”
In addition to current regional shortages, it is projected that the San Francisco Bay Area, Central Valley, and Central Coast regions of California will face substantial shortages of nurses by 2035.
Similarly, a 2018 study in the American Journal of Medical Quality forecasted that California will face the largest registered nurse (RN) shortage of any state by 2030, a shortage of more than 141,000 RNs. (For readers unfamiliar with the various nursing levels, visit here for a hierarchy of nursing.) The smaller-than-optimal number of new graduates is no accident.
The California Board of Registered Nursing (CBRN), which operates much like a medieval guild, limits the number of students who can attend nursing school. The nurse shortage, therefore, is not a case of individuals electing not to enter the field—nor is it the case that not enough people meet the high standards that the field demands. To the contrary, according to the CBRN’s own reporting, more than twenty-one thousand qualified nursing students were denied enrollment during 2016−2017, or 60 percent of all qualified applicants. Only 4 percent of the nursing programs that enrolled fewer students than the previous year said that the decline was driven by a lack of qualified applicants.
Facing a statewide shortage of nurses and a pandemic, the CBRN kept enrollment caps in place, a morally reprehensible act. Professional cartels prevent training and employment levels from adjusting to market demands based on price signals. Markets would operate properly if allowed to function unencumbered by outside impediments.
Addressing shortages of nurses and other healthcare workers: key belated reforms
To remedy the nurse shortage, California state assemblymember Blanca Rubio (D−West Covina) introduced AB 1364 in 2019, which would have exempted nursing schools that meet certain criteria from some of the requirements set by the CBRN and removed other regulatory roadblocks to more RNs. In January 2020, just before lockdowns began, the bill died in committee.
On March 23, 2020, Gov. Newsom admitted that “our staffing is going to require more flex, it’s going to require more capacity as it relates to existing ratios, as it relates to current scope of practice. We’re going to have to do more on rules and regulations.” Another path to helping alleviate the shortage of nurses, therefore, is to expand the permissible actions of existing healthcare workers.
In the state of California, as in other states, scope-of-practice rules limit the tasks that specific healthcare workers can perform. The rules—a mixture of laws enacted by the state legislature and regulations adopted by government agencies—dictate which tasks nurses, nurse practitioners (NPs), physician assistants (PAs), and other healthcare workers legally can perform.
After completing school and exam requirements, healthcare workers receive their licenses from the state for a specific occupation that has scope-of-practice rules. For example, in California, NPs must operate under the direct supervision of a physician—a rule they have been fighting against for years. NPs can dispense medications after a physician’s order, but they may not dispense drugs in a pharmacy. The California Medical Association, which lobbies on behalf of physicians, argues that NPs lack the skills necessary to operate independently of a licensed doctor. Nurse practitioners disagree. Current law permits no more than four NPs to serve under the supervision of one physician—a 4-to-1 maximum staffing ratio.
Union contracts also restrict flexible assignment of workers. Carmela Coyle of the California Hospital Association, commenting on the need for more specialty ICU nurses, told the Sacramento Bee, “Is there some cross-staffing that can go on? There is some, but at the end of the day, we are in need of critical care nurses, those ICU nurses, and it is difficult and, in some cases, against union contracts to substitute other nurses into those areas.”
By loosening scope-of-practice rules, healthcare employers would gain flexibility in allocating workers, delegating responsibilities, and prioritizing care, while ensuring safety for their patients.
A week after Newsom’s March 23 admission above, he signed Executive Order N-39-20 that eased scope-of-practice rules and relaxed licensing, certification, and staff-to-patient requirements. It removed the cap on the number of NPs and PAs who can serve under a supervising physician (but it maintained the physician-supervision requirement). And some hospitals petitioned state regulators to ease rules that require certain beds to be set aside for certain types of patients. Those emergency liberalizations expanded the supply of workers and increased the ability of supervisors to optimally allocate workers and other resources as needed.
California officials also streamlined the process for reinstating inactive or retired healthcare professionals who have been inactive or retired for five years or less. To help recruit and deploy retired doctors and nurses, newly graduated nurses and other healthcare workers, the state of California created the California Health Corps. When it was unveiled, Gov. Newsom said, “To treat the rising number of patients with COVID-19, our state needs more workers in the healthcare field to join the fight. If you have a background in healthcare, we need your help. Sign up at healthcorps.ca.gov.”
The website is a centralized registry to help increase the number of healthcare workers that could be mobilized quickly, even offering to pay for malpractice insurance. But Health Corps was a flop. Of the ninety-three thousand people who signed up, less than 1 percent were ever ready for deployment as of December 2020. Most people who registered were not qualified.
Desperate for more nurses in the workforce, the state also issued waivers to allow nearly nine thousand nursing students to graduate early by completing half of the clinical training required online instead of in hospitals with patients. Time spent working during the crisis also counted toward their clinical hours. In San Francisco, it normally takes an extraordinary six months to hire one nurse because of city regulations. The city waived some rules to allow administrators to hire nurses on the spot, a freedom that should have been granted long ago.
Earlier, the Proclamation of State of Emergency on March 4, 2020, allowed for the suspension of some licensing requirements for hospitals (visit here for a list of state licensing and certification waivers covering hospitals). The emergency proclamation also permitted the hiring of out-of-state medical personnel: “Any out-of-state personnel, including, but not limited to, medical personnel, entering California to assist in preparing for, responding to, mitigating the effects of, and recovering from COVID-19 shall be permitted to provide services in the same manner as prescribed in Government Code section 179.5, with respect to licensing and certification.”
The California Emergency Medical Services Authority, however, must approve out-of-state hires in advance after receiving a completed form for each and every prospective new hire, a slow process. On December 7, 2020, Gov. Newsom announced that the state would hire 435 temporary ICU nurses, including some from outside California, to combat the winter surge in COVID-19 hospitalizations and ICU admissions. Newsom said, “I remind you the issue at hand for us, the primary issue today and likely for the upcoming future ... is staffing.”
On March 12, 2020, Executive Order N-25-20 allowed the director of the Emergency Medical Services Authority to expand local EMS scopes of practice without consulting local EMS regulatory committees. The order also suspended the certification and licensure requirements for lab personnel to test samples for the coronavirus, provided that the person meets the Clinical Laboratory Improvement Amendments’ (CLIA) requirements, and the test is performed in a certified public health lab or licensed clinical lab.
On March 21, 2020, Executive Order N-35-20 allowed the director of the CDPH to waive many licensing and staffing requirements covering clinics, adult day healthcare centers, hospices, and mobile healthcare units.
On December 11, 2020, the Newsom Administration unveiled a fast-track waiver process for hospitals to increase the number of patients that a nurse can treat at one time. For ICU nurses, the number increased to three patients from two.
Ironically, Newsom’s Executive Order N-33-20, which discouraged preventative and non–life-threatening healthcare services, coupled with the lack of PPE, caused many healthcare workers to be furloughed. As University of California, Berkeley, researchers reported,
Between March 15 and July 11, 2020, approximately 251,100 healthcare workers in California filed an initial Unemployment Insurance (UI) claim and were paid benefits. Of these workers, 191,500 worked in ambulatory care services, 33,800 worked in hospitals, and 25,800 worked in nursing and residential care facilities. Aside from the foregone healthcare to patients and the income loss to many of these workers, the total budgetary cost of these unemployment claims is quite large: although claims details have not been published, we estimate that it is likely that these healthcare workers have received hundreds of millions of dollars in Unemployment Insurance benefits since the pandemic began.
Such circumstances highlight the inconsistency of state policies. On the one hand, Executive Orders N-35-20 and N-39-20 increased workforce capacity, while, on the other hand, Executive Order N-33-20 caused some healthcare workers to be laid off. Some layoffs could have been prevented with proper levels of stockpiled equipment, or sufficiently responsive supply chains to deliver needed supplies.
Nursing is not the only healthcare occupation in California experiencing shortages
Other healthcare occupations experienced shortages pre-COVID that worsened after the outbreak. Those occupations include physicians and respiratory therapists (RTs). The root causes are the same as with nurses: professional cartels restrict entry severely and government regulations reinforce the entry restrictions, limiting flexibility when needed most during a crisis. These arrangements benefit incumbents in the form of more generous compensation, but they hurt consumers and impede markets from conveying accurate signals to best allocate workers who have the needed skills.
California has experienced a chronic shortage of RTs before COVID, which became worse after COVID struck. RTs are medical workers trained for two years regarding how to operate ventilators and provide care for patients on ventilators or people with breathing disorders. RTs are licensed in every state and have taken center stage in the fight to save critically ill COVID patients. “I could not even tell you what a respiratory therapist does, exactly. But I can tell you that we’d all be toast without one on hand, especially with COVID patients,” said a California Pacific Medical Center nurse in San Francisco.
Using data from the ACA Annual Hospital Survey of FY 2018 and data from the Institute for Health Metrics and Evaluation COVID-19 Health Care Utilization Projections, the Fitzhugh Mullan Institute for Health Workforce Equity at George Washington University estimates that California needs to add 174 RTs for an adequate response to COVID-19.
With the increased demand for RTs to treat COVID patients, California does not have a sufficient supply of RTs for non-COVID patients. At peak hospital utilization, more than 100 percent of the state’s RTs would be needed to treat COVID-19 patients.
The Fitzhugh Mullan Institute also found that California requires 1,321 additional intensivists to respond adequately to COVID-19. Intensivists—also known as critical care physicians—are board-certified physicians who provide specialty care for critically ill patients, usually in intensive care units. The occupation acquired a new level of importance and surged in demand after the outbreak. Moreover, California has a shortage of 555 hospitalists, physicians whose primary focus is treating patients inside hospitals.
Overall, before COVID, California had a shortage of 4,100 physicians and 600,000 home healthcare workers, according to a 2019 report by the Future Health Workforce Commission. The California Health Care Foundation estimated pre-COVID that California will need 10,500 more primary care physicians by 2030 to treat its population properly.
The shortages have increased since COVID hit the Golden State because of a surge in demand and because thousands of medical workers are at home due to self-isolation after possible exposure to the virus. As of November 30, 2020, more than fifty-two thousand healthcare workers in California have contracted COVID-19, killing 218, since the start of the pandemic.
It requires many years of schooling and training to become a physician—eleven steps in the United States. But, as discussed in the recommendations section, reforms would increase the supply of physicians and other healthcare workers without compromising the quality of care. In fact, patient care would be enhanced.
Summary
Regulations created an enormous shortage of healthcare workers in California before COVID-19. California would have thousands more healthcare workers today to handle the surge of COVID patients if governments had not maintained artificial restrictions on the healthcare labor force for decades. The outbreak exacerbated the shortages, which were avoidable if California never had imposed labor regulations and instead allowed markets to guide education, training, and hiring decisions.
The thicket of rules prevented a fast and flexible response to the crisis by quickly hiring more workers, hiring workers from outside the state, hiring workers from outside the country, increasing the number of patients for whom workers are permitted to care, giving qualified workers more independence, moving qualified employees to other jobs as needed, or a mixture of such responses. Government rules blocked efficient responses to new conditions. Healthcare consumers paid a heavy price both before COVID and during the outbreak.
After liberalizations eventually were adopted, qualified healthcare workers from out of state were allowed to work in California (albeit with approval from the government after filing paperwork). Retired workers were welcomed back. Scope-of-practice restrictions and staffing ratios were relaxed, giving supervisors more flexibility. Licensing requirements were eased to allow faster hiring of healthcare personnel and more allocative flexibility. Those were sweeping improvements.
The emergency liberalizations need to be continued in perpetuity in order to help Californians, pandemic or no pandemic, especially low-income Californians living in communities underserved by healthcare facilities. If it is the right thing to do now, it is the right thing to continue doing in the future.
IV. Botched COVID-19 Testing
The most glaring governmental failure during the pandemic has been in the area of testing. Testing is crucial for getting a handle on the coronavirus outbreak by identifying infected people, tracing and testing contacts to contain spread, and ensuring that people infected by the virus are isolated quickly and treatments started promptly. Testing for antibodies can also help identify people who recovered from infection, perhaps while not exhibiting symptoms.
Many public health experts contend that the coronavirus could have been crushed by late spring or early summer 2020 if the FDA had permitted distribution of an inexpensive, nonprescription, at-home, self-administered test with rapid results in minutes. By performing 20 to 40 million at-home tests daily in the United States at a frequency of two times per week for each individual, it would have been possible to identify the status of everyone, isolate people who test positive, and focus restrictions on infected people, not on everyone. Lockdowns, masks, and social distancing might have been avoided or at least lessened (watch a discussion here). Personal risks could have been assessed more accurately soon after the emergence of the coronavirus. Instead, the federal government derailed that approach.
The federal government botched the initial rollout of test kits, required use of test kits that the federal government developed from scratch, and required central lab-based analysis. Later it allowed private tests but required a prescription to access such tests and required that the tests be administered by CLIA-certified laboratories and be nearly 100 percent accurate (yet, ironically, the FDA’s standard for authorizing vaccines was a 50 percent effectiveness rate). The government chose to mop up the mess using lockdowns and massive “stimulus” programs, instead of turning off the spigot relatively cheaply and quickly with frequent testing—frequency is key.
On February 7, 2020, the CDC began sending diagnostic kits to local public health laboratories, kits that the CDC designed. But many of the test kits did not work properly. As late as July 2020, Robert Redfield, then CDC director, blamed the problem on a manufacturing glitch. But according to an internal CDC review, obtained by National Public Radio and reported on November 6, 2020, poor quality control in the laboratory was the true culprit for the faulty tests, not a manufacturing problem. Even more troubling is that the CDC knew the kits failed 33 percent of the time, yet it allowed the kits to be distributed anyway without notifying recipients and instead of recalling the kits. All of those problems delayed the start of reliable testing nationwide by more than a month, until March, a critical loss of time that forced people to fly blind as fear increased.
At the same time, private laboratories were prepared to issue test kits but were blocked by the FDA. In Seattle, a project sponsored by the Bill and Melinda Gates Foundation was ready to test, but in a move labeled “bizarre” by a distinguished scientist, the FDA shut down the project pending more data. Not only is the FDA involved in testing oversight, but the CMS also imposes a gauntlet of regulations that must be satisfied before a new test can be used on the public.
Dr. Helen Chu, one of the researchers involved in the Seattle-based center, had been collecting swabs from symptomatic persons for an unrelated flu study. When she tried to repurpose her team’s flu test for COVID-19 infection surveillance, she was unable to do so because the new COVID test lacked formal government approval. As reported by the New York Times, “The Seattle Flu Study illustrates how existing regulations and red tape—sometimes designed to protect privacy and health—have impeded the rapid rollout of testing nationally, while other countries ramped up much earlier and faster. Faced with a public health emergency on a scale potentially not seen in a century, the United States has not responded nimbly.”
Though private-sector manufacturing and administration of tests would have expanded capacity considerably, the FDA mandated that testing kits be approved by them, stopping private actors from rolling out tests for about a month and a half.
Belated governmental responses that improved testing
Eventually, on March 16, 2020, the FDA reversed course, and its guidance statement included (1) a policy allowing states to assume oversight responsibility of laboratory-developed tests within their borders, with no FDA EUA required to deploy the test; (2) a policy for commercial manufacturers to distribute more rapidly new COVID-19 diagnostic tests to laboratories for use prior to the FDA granting an EUA, and (3) a policy for test innovators who are developing new serological tests and want them to be distributed and used without an EUA.
Those changes were important improvements, as they allowed for more testing in the United States, but valuable time had been lost, and prescriptions for lab-based polymerase chain reaction (PCR) testing with turnaround times of days, not minutes, made the tests, which were 100 percent accurate, also 100 percent useless from a public health perspective for containing the spread because the results took too long.
In partnership with a number of federal government agencies and reputable academic medical centers, manufacturers of tests could send samples voluntarily to verify testing accuracy. But on May 4, the policy was changed again to require commercial test developers to request Emergency Use Authorization with their validation data, a step backward from the FDA’s March 16 statement. Lab-developed tests created and used by a lab were not required to seek an FDA EUA if the state accepted responsibility for regulatory oversight of the lab.
All of these back-and-forth policies, often confusing and frequently changing, provided some needed capacity expansions. But it happened too slowly and not before the United States in its per capita testing levels, with headlining as one of the nation’s most visible examples of poor testing rollouts.
Along with the problem of deploying accurate tests quickly was the problem of having adequate laboratories to process the tests. The CMS, FDA, and state health departments each have roles in the certification, regulation, and accreditation of laboratories, whether public or private. Depending on the lab, one or more of the agencies determine whether and when a lab legally can process COVID tests. Many labs waited for government approval before they could process tests, losing more valuable time.
With a goal to achieve widespread testing and his back against a wall, Newsom ultimately turned to the private sector, announcing in August 2020 a $1.4 billion COVID-19 testing contract with Massachusetts-based PerkinElmer, which included a written guarantee that test results would be returned in one to two days. The state also has testing contracts with OptumServe and Verily, but neither of the contracts incorporates quick-turnaround guarantees—one to two days, however, is not quick enough for containing COVID’s spread.
As part of the state’s testing endeavor, the CDPH relied in part on the California Reportable Disease Information Exchange (CalREDIE), an electronic infectious disease reporting database. Test results and other information were to be accessible in real time and be available for health officials around the clock. But after hundreds of thousands of tests became backlogged, the goal of accurate real-time infectious surveillance was not achieved. In a survey issued to California’s fifty-eight counties, it was found that of the “thirty-three counties that provided information about test results, 94 percent said it often took more than two days to confirm results, while 58 percent of counties reported results taking as much as a week.”
Partially responsible for the slow processing of tests and the associated backlog were extensive technical difficulties in the CalREDIE system, which could not handle the large quantity of data, leading to data failures and backlogs. In mid-July, the CDPH switched to a tiered list to alleviate some of the bottlenecking. Testing delays were so long, however, that results were largely useless for isolating and effective contact tracing.
Dr. Mark Ghaly, secretary of California Health and Human Services said, “There’s a specific component that feeds information from labs to both the state’s system and the local public health system. That may actually be the place where data is getting stuck . . . . Absolutely, [contact tracing] will be hindered without this information.” When people receive test results after significant delays, they unknowingly can infect others as they await results.
Shortly after the CalREDIE technical difficulties were resolved, Dr. Sonia Angell resigned in August 2020 as director and state public health officer in the CDPH.
Returning to testing, anything less than nonprescription, inexpensive, rapid, at-home test kits, which facilitate frequent testing, will fail to crush the spread quickly. Unfortunately, as is standard practice, the FDA restricted direct-to-consumer at-home testing, allowing the virus to spread and lockdowns to be justified as the preferred governmental response.
Exactly one year after the first known human case of COVID-19, the FDA issued an EUA on November 17, 2020, for the Lucira COVID-19 All-In-One Test Kit, an at-home, self-administered test kit. The kit provides test results quickly, but is available only by prescription from a licensed physician, a barrier that will render the test effectively useless for containing the spread since most people will not take the additional time or pay additional money to visit a physician first. Another prescription-based, at-home test was authorized on December 16, 2020, Abbott’s BinaxNOW COVID-19 Ag Card home test.
On December 15, 2020, the FDA issued an EUA for a nonprescription COVID-19 rapid home antigen test by Ellume, an Australian company. It was the first over-the-counter (OTC), fully at-home diagnostic test to receive authorization. According to the EUA, “Results are delivered in as little as twenty minutes to individuals via their smartphone. ... Ellume expects to produce more than three million tests in January 2021.” That is an important step toward comprehensive testing, but three million tests and a gradual rollout is too little, too late. And the $30 price tag—less than Lucira’s $50 price tag plus the cost of a doctor’s visit—makes mass testing twice a week unaffordable for many people (another strong argument for radical changes in the patent system, as discussed below in the recommendations section).
The slow authorization of rapid OTC, at-home tests has been a costly mistake, contributing to unnecessary deaths, say epidemiologists. Dr. Peter Chin-Hong, a professor of medicine and infectious diseases at UCSF, said, “Everything is too late, unfortunately. We should have had [rapid home] testing long ago. Lack of testing is one of the main reasons why we are here, with the surge and lack of hospital beds.” Chin-Hong added that rapid tests could have been approved months ago: “We’ve had this technology forever. This is like basic stuff if you talk to the scientists.”
In contrast, the slow PCR tests administered by healthcare professionals (for example at Dodger Stadium in Los Angeles) and analyzed using sophisticated sofa-sized PCR machines in high-tech laboratories are useless from a containment perspective. Dr. Michael Mina, an assistant professor of epidemiology at Harvard University, said, “We’re starting to see four- or five-, six-, seven-day delays, which frankly makes these high-quality PCR laboratory tests complete garbage, completely useless. ... If you’re waiting for five days to get a test result back, it’s not even worth getting the test.” Yet, this has been the approach favored by government officials for almost a year.
Summary
Early in the COVID-19 outbreak, it became clear that communities needed rapid, inexpensive, frequent testing. Yet procuring tests was a disaster from the start. The CDC botched the initial rollout and covered up its mistakes, while, at the same time, the FDA prevented private alternatives from reaching the market. Only after opening up the testing market to private innovators and more laboratories was progress made; yet the goal of rapid and frequent, at-home, nonprescription testing has not been achieved after one year—an unmitigated disaster that has cost lives and devastated the economy because of government-imposed lockdowns being the preferred response.
Contrast the heavy-handed, micromanaged, botched approach for developing tests with the lighter-touch and accelerated approach used to develop vaccines and therapeutics—the subject of the next section.
V. Vaccine and Therapeutic Antiviral Drug Development
In a public health environment where people are experiencing unexpected infections and deaths, it is understandable that they want effective vaccines and therapeutics as soon as possible. For definitional purposes, according to the CDC, a vaccine is a “product that stimulates a person’s immune system to produce immunity to a specific disease,” protecting the person from acquiring that disease. A therapeutic antiviral drug treats viral infections in people already infected by inhibiting replication of the virus.
Dr. Soumya Swaminathan, an epidemiologist with WHO, noted, “[T]he SARS-CoV-2 virus is a highly transmissible virus. We think it needs at least 60 to 70 percent of the population to have immunity to really break the chain of transmission.” Since vaccines can accelerate immunity, the motivation is clear to quickly develop a safe and effective vaccine. But typically, vaccines take more than ten years to develop. The mumps vaccine, which took four years to develop, held the record for fastest development prior to COVID-19.
The key governmental response: Operation Warp Speed
On May 15, 2020, about two months after WHO declared a pandemic, the Trump Administration unveiled a fast-track vaccine program called Operation Warp Speed (OWS), a Manhattan Project–style effort to dramatically cut the time needed to develop an effective coronavirus vaccine. OWS and the National Institutes of Health’s coordinated research strategy program called Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) are public-private arrangements involving the Department of Health and Human Services (HHS), Biomedical Advanced Research and Development Authority, CDC, and FDA, among others. Operation Warp Speed consists of separate teams tracking vaccine development and providing logistical support for procuring and mobilizing vaccines.
The stated goal of OWS is to “produce and deliver 300 million doses of safe and effective vaccines with the initial doses available by January 2021, as part of a broader strategy to accelerate the development, manufacturing, and distribution of COVID-19 vaccines, therapeutics, and diagnostics (collectively known as countermeasures).” OWS is intended to ensure that a handful of companies that are developing promising potential vaccines do not encounter “technical, logistic, or financial hurdles” that “hinder vaccine development or deployment.”
OWS support has often—but not always—involved a significant upfront financial investment by the federal government. Through the Coronavirus Aid, Relief, and Economic Security (CARES) Act, $10 billion has been appropriated for the research and development effort. Essentially, the federal government is placing bets on a portfolio of promising approaches, hoping that one or more will be successful and can be distributed rapidly to the public. It has been called a “moon shot” approach to vaccine development and distribution.
Five companies received OWS assistance for their vaccine candidates starting in June 2020: Moderna, Astrazeneca (partnering with Oxford University), Janssen Pharmaceutical Companies of Johnson & Johnson, Merck, and Pfizer (working in partnership with German biotechnology company BioNTech). Although Pfizer financed its research and development independently of OWS, it agreed to distribute at least 100 million doses of its vaccine, if safe and effective, to the US government for $2 billion under an OWS advanced purchase agreement.
In early July, the federal government also agreed to invest $1.6 billion in Novavax’s vaccine, through clinical trials and manufacturing of 100 million doses in the United States. In late July, Sanofi, in partnership with GlaxoSmithKline, was granted up to $2.1 billion to speed up its clinical development and manufacturing of the pair’s vaccine candidate. The initial order is for 100 million doses for the United States. The federal government also invested $628 million in Emergent BioSolutions to expand vaccine manufacturing capacity in the United States. Inovio’s vaccine and Vaxart’s oral tablet vaccine also were selected for a nonhuman primate “challenge” study funded by OWS. In such a study, a vaccine’s evaluation is fast-tracked by immunizing healthy participants and then deliberately infecting them with the virus.
Finally, OWS spent less than $1 billion on the development and manufacturing of three monoclonal antibody treatments for people infected with the coronavirus. The two candidates that are furthest along are from Regeneron and Eli Lilly. Regeneron’s drug was administered to President Trump after his COVID-19 infection.
On November 9, 2020, the FDA issued an Emergency Use Authorization for Eli Lilly’s monoclonal antibody therapy to treat mild to moderate coronavirus infections in adults and children who are not hospitalized. It is the first authorized use of a monoclonal antibody in treating COVID-19, designed to jump-start the body’s immune response against the infection by attacking the spike protein of the coronavirus. Lilly entered into an advanced purchase agreement with the federal government for three hundred thousand doses to be delivered within two months of the EUA, and it expects to manufacture a million doses by the end of 2020. Regeneron Pharmaceuticals also received an EUA for its monoclonal antibodies cocktail, called REGN-COV2, on November 21, 2020.
Both monoclonal therapies, however, suffer from regulatory and logistical problems that make them difficult to administer effectively. For example, both drugs can be administered only to patients not hospitalized, an FDA regulation that one prominent scientist called “completely silly.” Dozens of antibody therapies are in various stages of trials and investigations, which have been accelerated through ACTIV.
The National Institutes of Health operates ACTIV, which is intended to speed up the evaluation of candidates in trials by advising on the proper “protocol designs and endpoints to ensure a harmonized approach across multiple vaccine efficacy trials” being implemented under OWS. The initiative coordinates clinical trials by using common institutional review boards, safety monitors, and protocols. Regarding the relationship between ACTIV and OWS, a Warp Speed official said that “no conflict [exists] at all—they are working together—one intellectually (ACTIV) and one operationally (Warp Speed).”
Other factors facilitated development of COVID-related drugs, such as “the-whole-world-is-watching” effect and competition among countries to be first to authorize deployment of effective vaccines and other COVID drugs. Normally, FDA actions are not followed daily by the public. Also, FDA actions are often guided by the “precautionary principle,” which makes agency officials highly risk averse, causing the agency not to approve drugs or to approve drugs very slowly because of excessive bureaucratic red tape.
But as noted by economists William F. Shughart and Pierre Lemieux, “Precaution can kill. Especially, we should add, in times of pandemic. Under the precautionary principle, we’ve put inventors and innovators in an impossible situation. They have to show that their products will have no harmful consequences—ever. The benefit side of the ledger is ignored. In the meantime, inventors stop inventing. Persons suffer as a result, possibly even dying, from maladies the innovators could have helped them to avoid.” That has been less the case with COVID-19 drug development because many countries wanted to be first to authorize use of a vaccine, and people were watching FDA actions everyday, which point to needed reforms that are discussed in the recommendations section below.
Rather than the federal government providing OWS money to companies by issuing contracts directly to them, more than $6 billion in funding was routed through defense contract management firm Advanced Technologies International. That firm assumed the responsibility of awarding contracts to companies.
One of the advantages of using Advanced Technologies is expediency. Government contracts of the same nature typically would be subject to the Federal Acquisition Regulation in Chapter 1 of Title 48 of the Code of Federal Regulations. It governs solicitations and contract clauses, leading to potential delays, especially if multiple bidders compete or if one of the losing parties submits a protest.
In exigent circumstances, the government’s streamlined alternative, used by Warp Speed, is the Other Transaction Agreement (OTA), which allows the government to use a nongovernmental organization as an intermediary to bypass regulatory oversight and transparency requirements. Standards and practices are weaker with OTAs.
A 2019 Congressional Research Service report warned that OTAs come with “diminished oversight and exemption from laws and regulations designed to protect government and taxpayer interests.” In 2018, Scott Amey, the general counsel for the Project on Government Oversight warned, “We have to seriously consider how we are using [OTAs]; whether we are using them as intended, whether we are getting the goods and services that we really want and need, whether we are getting them at the best cost and prices, and [whether] we are using this procurement vehicle as a way just to circumvent the rules and have contractors not have the administration and oversight they need to hold them accountable. I’m just afraid this is going to result in a lot of waste, fraud, and abuse in the future.”
After vaccine company Novavax announced $1.6 billion in funding from OWS, Sydney Lupkin, a reporter with National Public Radio, and Kathryn Ardizzone, a lawyer at Knowledge Ecology International, submitted public records requests to the US Department of Health and Human Services. Responses to the requests uncovered that the government had no record of the contract. Ardizzone said that it was unclear if using the Freedom of Information Act would reveal the agreements since they are maintained by a private entity, Advanced Technologies. Without transparency, no one can know if taxpayer protections and other procurement standards have been included in the agreements.
Another concern with OWS is the omission of potential foreign vaccine developers. In a memorandum to the COVID Task Force, Peter Navarro, then director of the Office of Trade and Manufacturing Policy, stated that fast-tracking efforts should prioritize US-based vaccine companies, without justifying that preference. It would seem irrelevant where a vaccine producer is located provided that it is allowed to sell anywhere in the world to whomever wants to buy the vaccine. Any company that successfully develops a vaccine likely will have the capacity to distribute the vaccine globally since assistance is offered by many governments.
An unnamed federal government scientist who is involved in OWS stated that although Warp Speed has not ruled out any specific type of vaccine [platform], Science magazine reported that OWS “will not consider ones made in China, such as the inactivated virus vaccine recently shown to protect monkeys from the coronavirus, a first.” “We can’t partner with Chinese companies. That’s just not going to happen,” the unnamed scientist said.
Seth Berkley, who heads Gavi, a global alliance for vaccines and immunizations, said that the decision to exclude Chinese products is shortsighted, given that China was a few months ahead of the United States in exploring vaccine options. By mid-fall 2020, several of the leading vaccine candidates were developed by companies headquartered in China, including the company Sinovac, whose vaccine candidate was found to be safe, and CanSino Biologics, whose vaccine was approved for the Chinese military in June 2020.
Summary
The most important success story to emerge from the COVID-19 pandemic has been the fast development of several vaccines (hopefully widely effective). It is important to note that this happened by unleashing the innovative potential of nongovernmental research entities—most of them private pharmaceutical companies—while at the same time minimizing government interference in the creative process. In fact, Pfizer explicitly rejected US government research grants in order to maintain its independence and “keep Pfizer out of politics.”
Advanced purchase agreements between the federal government and vaccine companies guaranteed minimum payoffs for any successful vaccine, which reduced the companies’ risks. Predevelopment funding was expedited through OTAs. Clinical trials and FDA reviews of trial data were fast-tracked by ACTIV. Basically, the government tried to stay out of the way of scientific innovators to eliminate the stranglehold of bureaucracy that normally grips new drug development.
The strategy proved successful. On December 11, 2020, Pfizer’s COVID-19 vaccine became the first to receive FDA Emergency Use Authorization in the United States. Previously, on December 2, 2020, the United Kingdom became the first country to authorize its use. The Pfizer vaccine went from concept to approval in ten months. It normally takes ten years in the United States. Moderna’s vaccine received an FDA EUA on December 18, 2020. Moderna’s vaccine was designed just two days after the novel coronavirus’s genomic sequence was disclosed publicly, meaning, as the title of an article in New York magazine’s Intelligencer indicates, “we had the vaccine the whole time.” Johnson & Johnson’s vaccine received an FDA EUA on February 27, 2021.
For a variety of reasons, however, including a confusing tiered prioritization system and a clunky software platform called PrepMod, California has struggled to get its allotted vaccines into people’s arms. In fact, during the first four weeks of vaccine distribution, only Alabama, Georgia, and Virginia performed worse than California in vaccinating its population. California vaccinated only 2,468 people per 100,000 compared to the national average of 3,300.
With a goal to accelerate COVID-19 vaccinations, California signed contracts in mid-February 2021 with Blue Shield and Kaiser Permanente to run a centralized statewide vaccine distribution program. Blue Shield will: (1) manage a network of vaccination providers; (2) devise “incentive payments” for providers who operate efficiently; and (3) create a data platform to guide distribution of vaccine doses. Kaiser will manage at least two mass vaccination sites.
Despite the stumbling vaccine rollout, the new approach to vaccine development—or an even more liberal approach as discussed in the next section—has the potential to accelerate significantly new drug development in the future. The gains could be revolutionary. And, again, if this approach is the right thing to do now, it is the right thing to continue doing from now on.
VI. Patient-Centered Recommendations
During the first months of the COVID-19 outbreak, government healthcare regulators tried to do business as usual, but it quickly became apparent that regulations, designed to benefit incumbents, were causing a crisis in care that was becoming worse. Many of the problems, especially personnel shortages, were avoidable if the regulations never had been imposed.
By liberalizing long-established rules and regulations, federal, state, and local government officials essentially admitted that the barriers and special privileges that had been created hurt consumers and made the delivery of healthcare services unworkable. With their backs against the wall, politicians and bureaucrats did the morally correct thing and began to loosen the regulatory stranglehold through sweeping reforms. The liberalizations should be made permanent after the crisis—the moral course of action.
Californians deserve a healthcare system that is quickly and nimbly able to respond to new circumstances; provides convenient, high-quality care at affordable prices; and encourages rapid innovation to fight emerging threats and offer lifesaving breakthroughs. COVID-19 revealed that regulations prevent consumers from benefiting from such developments and, moreover, that deregulation helps consumers without compromising safety.
The recommendations in this report fall into two categories. The first category is regulatory changes that already have been implemented on a temporary basis and, with the exception of the Defense Production Act and Operation Warp Speed, should be continued permanently or even expanded. The emergency liberalizations have been discussed in earlier sections of this report and are summarized in Table 1.
The second category is regulatory changes that have not yet been fully implemented but should be adopted in order to provide the best access to high-quality and cutting-edge healthcare at affordable prices for all consumers.
| Table 1. Emergency Liberalizations Resulting from COVID-19 That Benefited California Healthcare Consumers and/or Medical Personnel | |
| Personal Protective Equipment (PPE) and Medical Equipment | FDA authorized, in multiple EUAs, the use of unapproved medical products and unapproved uses of approved products if the company certifies the product is FDA compliant. Products include PPE, medical devices, diagnostic tests, ventilators, and more. |
| FDA allowed unlicensed companies to manufacture PPE and medical equipment. | |
| FDA fast-tracked the importation of PPE and medical devices subject to an EUA. These new rules were implemented by CBP. | |
| Trump Administration activated the Defense Production Act to compel production of medical equipment, to prevent the exportation of PPE, and to mobilize the military for COVID assistance. | |
| Healthcare Personnel | State scope-of-practice rules eased. |
| State licensing, certification, training, and staffing ratios relaxed for healthcare occupations and facilities. | |
| State rules streamlined to more quickly hire inactive/retired healthcare professionals and qualified out-of-state medical personnel. | |
| Some state training rules for nursing students waived, allowing the students to graduate early and be hired sooner. | |
| COVID-19 Diagnostic Testing | FDA reversed course, allowing commercial manufacturers to sell tests to laboratories without an FDA EUA (this was later reversed). |
| Test manufacturers could voluntarily send samples to government and academic labs to verify the accuracy of the manufacturer’s test (later reversed). | |
| FDA transferred to states oversight responsibility for lab-developed tests. | |
| Newsom Administration turned to the private sector and signed mass-testing contracts with OptumServe, PerkinElmer, and Verily. | |
| FDA issued EUAs for at-home, self-administered test kits developed by Lucira and Abbott (prescription required) and by Ellume (OTC). | |
| Vaccines and Therapeutic Drugs | Predevelopment and postdevelopment government funding of promising vaccines and therapeutics through Operation Warp Speed. |
| Channeled payments through a private contractor to expedite the process (OTAs). | |
| Expedited clinical trials and the review of trial data through the ACTIV program. | |
| FDA issued EUAs for monoclonal antibody therapies and COVID-19 vaccines. | |
| Source: The table was compiled by the authors using material in Lawrence J. McQuillan, Jonathan Hofer, and Douglas E. Koehler, COVID in California: How Government Regulations Created Critical Healthcare Shortages—Permanent Patient-Centered Reforms Needed (Independent Institute, April 2021). | |
Next, seven reforms are discussed that would help patients significantly:
1. Allow private companies in the United States and abroad to supply the equipment demanded by the healthcare sector.
Healthcare providers should be able to source needed products from private manufacturers in the United States or around the world without licenses or permits as long as each manufacturer certifies that its products are FDA compliant or compliant with the standards of a private governing body, much like Underwriters Laboratories certifies the safety of electronic equipment.
The default should be “permissionless innovation and competition,” with any damages stemming from problematic products being remedied in the civil courts through tort proceedings. The changes will help to ensure a reliable supply of needed equipment at competitive prices, crisis or no crisis. Protection of incumbents should become a relic of the past.
2. Abolish scope-of-practice rules and staffing ratios.
All scope-of-practice rules and staffing ratios should be eliminated so that healthcare administrators and supervisors in healthcare facilities decide who performs which tasks. Scope of practice should be determined at the point of care. It is absurd that lawmakers and bureaucrats are making such decisions by mandating which occupations can perform which tasks and dictating patient loads when healthcare supervisors are in the best position to judge the capabilities, training, and skills of specific employees and to determine where assistance is most needed. Eliminating such rules would help restore the flexibility that healthcare facilities need to care properly for patients and assign workers efficiently. A facility should be held accountable in civil court for any damages due to negligence, although no facility has an incentive to harm its patients.
Research demonstrates that adverse health outcomes have not occurred when states relaxed their scope-of-practice rules in the past. For example, a study in Medical Care Research and Review found no differences in health status between patient groups assigned to a nurse practitioner versus a primary care physician two years after treatment. Another study published in 2018 in the Journal of Health Economics found that health outcomes were comparatively better after scope-of-practice laws were eased.
On March 11, 2020, the Florida legislature passed House Bill 607, which was introduced in 2019. Quickly signed by Gov. Ron DeSantis (R), the bill expanded the scopes of practice for several categories of direct care workers. The language of the bill authorized personnel to perform additional duties that they were not previously permitted to complete at their level, but otherwise were capable of performing. Registered nurses, certified nursing assistants (CNAs), and advanced practice registered nurses (APRNs) achieved much independence as a result of the law.
The expanded scope-of-practice rules for APRNs meant that an RN could “delegate tasks, including the administration of medications, except controlled substances, to a CNA or HHA [home health aide] for a patient of a home health agency if the RN determines that the CNA or HHA is competent to perform the task.” California needs that level of flexibility, and more, on a permanent basis such that supervisors in healthcare facilities make scope-of-practice decisions, not government officials beholden to professional cartels seeking protection from competition.
Similarly, staffing ratios should be determined by supervisors in healthcare facilities on a worker-by-worker basis, not by government edicts. No two people are identical; therefore, patient loads should be determined by worker capabilities, not fixed formulas. Furthermore, each physician should supervise the number of NPs and PAs that they can supervise competently. Such workplace decisions should be left to supervisors, subject to the terms of any union contracts, not by government-imposed staffing ratios.
3. Reduce practice barriers for nursing and all other healthcare occupations.
It is impermissible and frankly immoral that during a nurse shortage the California Board of Registered Nursing maintained its nursing program admissions caps. With regional nursing shortages occurring throughout California for years, and stresses exacerbated during the pandemic, qualified students should not be turned away. The CBRN should not have the authority to impose admission limits, nor should any other professional cartel for any other healthcare occupation.
That recommendation points to the larger problem that excessive self-regulation buttressed by government rules backed by force has created practice barriers that increase the incomes of incumbents at the expense of affordable access to care for healthcare consumers. COVID-19 brought that unfair and immoral system to the surface.
Government occupational licensure of healthcare workers should be abolished and replaced by private third-party certification of workers. A white paper prepared by the Department of the Treasury, Council of Economic Advisers, and Department of Labor during the Obama Administration, concluded, “Licensing laws also lead to higher prices for goods and services, with research showing effects on prices of between 3 and 16 percent. Moreover, in a number of other studies, licensing did not increase the quality of goods and services, suggesting that consumers are sometimes paying higher prices without getting improved goods or services.”
Government occupational licensure of the healthcare industry is not needed, and evidence that occupational licensing in healthcare results in better health outcomes is spurious.
One alternative approach would be to sunset government-controlled occupational licensing and replace it with a system of private third-party certification (one possible variant is discussed here). Over time, trusted brands for certification would emerge, much as Underwriters Laboratories has become the gold standard of safety certification for many consumer products. Healthcare employers would hire from pools of applicants certified by trusted brands, and the best of them would be promoted. Individuals could expand their skill sets as their interests and market demands change by acquiring certifications over time (called “credential stacking”) from private credentialing organizations. Individuals could acquire credentials to perform specific procedures, and employers would judge their competence before permitting them to perform the procedure at a point-of-care facility.
Private third-party certification would allow the supply of healthcare workers and their skill sets to better match market demands (called “right skilling”) rather than being captive to professional cartels that use government mandates to stifle competition and block entry. Private third-party certification also would allow new fields of care to emerge organically as entrepreneurs observe occupational gaps and develop certification programs to meet the need. A private approach would reduce cartel and government influences and increase consumer-driven responses based on market signals.
Our credentialing recommendations apply to all healthcare occupations.
4. Permit interstate and international competition in healthcare occupations by allowing a freer flow of talent.
Consumers benefit from rigorous competition among providers and from having wider choices among providers. California should open its market to qualified healthcare workers in other states. During the COVID-19 crisis, California has allowed out-of-state hires, with strings attached. That option should become permanent and the strings cut. It should encourage providers to work in their chosen field in California, where shortages of healthcare workers across occupations and locations are growing, especially in rural California.
At the very least, California ought to become a member of healthcare professional compacts such as the Enhanced Nurse Licensure Compact (eNLC), or the Recognition of EMS Personnel Licensure Interstate CompAct (REPLICA). Those compacts allow workers in good standing to practice in other member states. The REPLICA website states, “Qualified EMS professionals licensed in a ‘Home State’ would be extended a ‘Privilege To Practice’ in remote states for qualified circumstances.” Nurses and other professionals who are qualified and credentialed in other states should be allowed to practice in California. (States likewise could form compacts that recognize certifications by other states of medical devices, a step toward the first recommendation above.)
Finally, foreign-born physicians in good standing should be allowed to practice medicine in the United States, with full disclosure of their credentials. Today, many skilled and experienced foreign doctors who make their way to the United States essentially must often start from scratch for certification (visit here for an excellent video on the topic).
Too many foreign-born physicians in the United States drive taxis, work at car washes, or manage supermarkets instead of saving lives only because cartel regulations and visa restrictions protect native US residents from competition from foreign-born doctors. The “brain waste” is staggering.
California needs more doctors, especially in rural areas and underserved communities. Allowing foreign doctors easily to practice in the United States and in California would give consumers more options—especially in their native languages and native cultures—and lower prices with more vigorous competition.
In California, where about 44 percent of the population speak a language other than English at home, a shortage of Spanish-speaking physicians is evident, according to a 2019 study by the Latino Policy and Politics Initiative at the University of California, Los Angeles. Replicating the streamlined approach adopted in Australia or Canada, as discussed by Paul Larkin Jr. here, would be a step forward in bringing more foreign-born physicians into the US healthcare workforce.
Brain waste exists in other healthcare occupations. The Migration Policy Institute estimated in April 2020 that California alone has sixty thousand immigrants or refugees with healthcare-related undergraduate degrees that are not being utilized fully. Researchers Jeanne Batalova and Michael Fix concluded that “these immigrants represent a potentially important source of staff for the US health corps. And because these immigrants tend to be younger than their US-born counterparts, they represent an important pool of responders to a disease [COVID-19] that is particularly dangerous for those sixty and older.”
Other liberalizations that would increase the supply of physicians in California, endorsed by the California Health Care Foundation, include (1) increasing the capacity of existing medical schools, (2) expanding the number of medical training programs, and (3) expanding residency programs.
High barriers to entry created and/or enforced by governments, such as limits on admissions to nursing schools, limits on residency slots, occupational licensure, and limits on the flow of talent across state lines and international borders, serve the special interests of incumbents, but harm patients.
5. Free up testing by allowing private companies to compete vigorously, providing tests, testing sites, and testing laboratories without government approval.
The state of California and the US government proved early on to be incapable of providing fast and reliable mass testing for COVID-19. As public outrage increased, government officials relaxed rules to encourage private-sector participation and innovation.
Rules should be changed to encourage nongovernmental entities to develop new tests, operate testing sites, and process tests in private laboratories. The gold standard, of course, are at-home, OTC test kits that are both inexpensive and rapid, allowing for frequent testing. Rivalrous competition among private companies can make this happen.
As with medical supplies, the standard should be permissionless innovation. Companies should be held accountable for fraud or damages through criminal or civil court proceedings. The same approach should become the norm for all testing, not just for COVID-19.
As with worker certification, private organizations, perhaps academic medical centers, will spot entrepreneurial opportunities to become the leaders in certifying the accuracy and reliability of tests and testing laboratories. Healthcare providers have a strong incentive to be duly diligent in identifying and adopting reliable tests, and in suing manufacturers of unreliable tests. The private sector should be encouraged to develop new tests using different platforms; administer those tests in private clinics, labs, or at home; and use private facilities to analyze the tests as needed to arrive at test results.
Because of the steady demand for testing, it is imperative that state and federal governments allow private companies to manufacture tests, private laboratories to analyze tests, and all to freely innovate.
The VITAL Act of 2020, sponsored by Sen. Rand Paul (R-KY), warned, “As witnessed with the 2020 COVID-19 pandemic, undue regulation of laboratory-developed testing procedures may hamper the medical management and public health response to infectious disease outbreaks and pandemics, leading to delays in access to testing and the ability to meet needed capacity to stem community spread.”
At the very least, California should borrow an idea in the VITAL Act, which is a guideline for regulatory boards to “ensure that regulatory oversight of laboratory tests does not limit patient access, impede innovation, constrain flexibility or adaptability, or limit a test’s sustainability as a result of being unduly burdensome or beyond the fiscal capacity of the laboratory to reasonably validate and perform.” In other words, regulation should not strangle the benefits of the thing it regulates.
6. Permanently allow telemedicine across state lines.
State licensing laws have interfered with the development of telemedicine because licenses restrict the practice of telemedicine only to those who are licensed in a state. That restriction has been lifted temporarily during the pandemic, allowing telemedicine across state lines. The restriction should be eliminated permanently, allowing telemedicine to flourish.
In California, the original Proclamation of a State of Emergency and Executive Order N-43-20 relaxed limits on the practice of telemedicine by in-state and out-of-state healthcare professionals. An HHS waiver relaxed rules at the federal level. California insurance commissioner Ricardo Lara directed health insurance companies to provide more access to telehealth services and provide reimbursement parity between telehealth and comparable in-person services. Similarly, a Medicaid waiver for California allowed for the reimbursement of payable claims by out-of-state telehealth providers. The CMS eased restrictions on telehealth in the Medicare Advantage program. A series of notices allowed audio-only telephone calls to deliver services, in addition to various video platforms.
It would be devastating to many people if those emergency waivers are ended. It is difficult for many people to visit a clinic or hospital for care, especially older persons, people with disabilities, and people without reliable transportation. Telemedicine has become a godsend for them.
Telemedicine is another argument for replacing state licensing with private certification. If a medical professional is certified as competent in an area of medicine, and a telemedicine company wants to hire him or her for patient care across state lines, they should be able to do so.
7. Speed the development and deployment of new vaccines and therapeutic drugs.
A number of reforms could be continued or adopted that would speed the development of new vaccines and therapeutic drugs. Ten years, on average, to develop a new vaccine is nonoptimal. The recommended changes range from small tweaks to radical new approaches that are worthy of further consideration.
- The federal government should continue and expand the ACTIV program to expedite clinical trials and to accelerate the review of trial data. In addition, the FDA should expand the evidence it considers during the drug approval process to include big data analysis and other approaches.
- The mandate of the FDA should be changed to require it to evaluate only the safety of a proposed new drug, not its efficacy (for a discussion see “The Case Against Patents” by Michele Boldrin and David K. Levine in the Journal of Economic Perspectives, 2013).
Such a change would ensure that dangerous drugs are kept off the market but would also allow safe drugs to be distributed to the public sooner so that people who could benefit from new drugs can obtain them sooner. The reform would help reduce the cost of new drug development, now running in the range of $1.3 billion to $2.8 billion per drug and taking up to a decade to see the light of day. - If the FDA’s mandate was scaled back to safety, not efficacy, then initial R&D costs would be reduced, and a strong case could then be made for shortening the duration of pharmaceutical patents. Patent holders are typically granted protection for a period up to twenty years from the date of filing. A reduction in the patent term would reduce the propensity to file frivolous patent applications with no intention to use the patent productively. Such a reform has the added benefit of reducing the total number of patents that other innovators must navigate to ensure they are not infringing on an existing right holder. Complex arrays of property rights, or “patent thickets,” can form, reducing the application of new ideas and making it more difficult to commercialize new products. If the FDA delays the approval of a given drug, a period of time can be tacked onto the duration of the patent equal to the FDA delay.
- Drugs approved in select foreign countries should be allowed on the US market, with appropriate disclosures. Reciprocity would permit drugs to reach consumers quicker in the United States, bypassing FDA regulatory bottlenecks. Reciprocity should apply to medical devices and PPE as well.
Both the Pfizer/BioNTech vaccine and the Oxford/AstraZeneca vaccine were authorized for use in the United Kingdom before they were authorized in the United States. If reciprocity existed, millions of Americans could have been immunized sooner, perhaps saving hundreds or thousands of lives. - Patents granted from applications dating before 1995 were not disclosed automatically. Patent applications could be kept secret during the application process if the applicant kept submitting continuation requests, which kept the patent pending until the applicant wanted to receive the patent and receive the full length of protection. The propensity for such patents to “surface” in legal disputes earned the practice the moniker “submarine patents.” Greater transparency in the application process has helped mitigate that problem. Now applications are published automatically eighteen months after the priority date; the Patent and Trademark Office publishes the application then whether or not a patent is awarded. If applications were disclosed immediately upon first filing, other innovators would be granted more time to review potential conflicts.
- Operation Warp Speed should be wound down, but its “prize” feature should be adopted widely in the United States and around the world. Under a prize model, governments, private nonprofits, and other entities could offer lump-sum grants—$5 billion, say—to a company or partnership that develops a drug that cures an identified disease such as the common cold, HIV-AIDS, or a form of cancer. The prize model was used, in effect, for the development of Pfizer’s COVID-19 vaccine, since the company rejected up-front US government money, but accepted a $2 billion advanced purchase agreement (the “prize”).
The prize model reduces government interference in research and development while freeing entrepreneurs to innovate. More drug advances potentially are achievable by adopting the same innovative approach going forward. (To learn more about the power of prizes to incentivize innovation, visit here, here, and here.) - A variation of the prize model would require the “winner” of the prize not to patent the new knowledge as a condition of receiving the prize. Such a stipulation would replace patent protection with a lucrative prize and allow the world’s scientific community immediately to learn from and utilize the new knowledge. Nobel laureate economist George Stigler was sympathetic to that approach, writing in 1968, “One could therefore argue that inventors should be rewarded by lump-sum grants rather than by exclusive ownership [patents] in order to permit the socially desirable unlimited use of knowledge.”
In the interim, calls are growing, led by India and South Africa, to waive patents and IP rights globally for COVID-related advances during the pandemic in order to distribute useful innovations as widely as possible. (A more extreme idea, worthy of further scholarly analysis, is to phase out statutory intellectual property (IP) protection entirely based on the argument that IP restricts the dissemination of new ideas, reducing innovation and human progress. Read “The Case Against Patents” for additional discourse and scholarly research on ending IP. Watch Michael Heller, a professor at Columbia Law School, explain how high transaction costs associated with patents, such as costs arising from patent thickets, can reduce pharmaceutical innovation.)
Patent protection is not necessary for an important new idea to make its way to market. Although it was discovered in an era when billion-dollar investments were not required to make significant advancements, antibiotic penicillin was one of the most important medical innovations during the past century and it was not patented.
In 1928, Scottish bacteriologist and physician Alexander Fleming discovered the antibacterial properties of a mold that contaminated one of his petri dishes in his laboratory. At the time, no cure was available for infectious bacterial diseases such as cholera, typhus, or pneumonia. As a result, the average life expectancy at the beginning of the twentieth century was forty-seven years in the industrialized world. Despite early successful experiments showing penicillin’s effectiveness in killing a few common forms of pathogenic bacteria, the antibiotic largely was unusable as a therapeutic treatment without purification. Fleming and his team experimented with purification, but to no avail, and “penicillin languished largely forgotten.”
Penicillin research resumed in 1939 at the University of Oxford by Australian pharmacologist Howard Florey, who received funding from the Rockefeller Foundation. As World War II broke out in Europe, a tremendous need for treating wounded soldiers experiencing high mortality rates from infections became evident. With British drug makers overburdened due to the ongoing war effort, Florey did not have the means to make enough of the drug for preliminary trials. After traveling to the United States, Florey was able to recruit American pharmaceutical companies Pfizer and Eli Lilly to help mass-produce purified penicillin. The companies succeeded to such an extent that when the United States entered World War II, the War Department considered penicillin manufacturing to be a high priority, second only to the Manhattan Project. Despite having the opportunity, Howard Florey refused to patent penicillin for ethical reasons. Now the “wonder drug” penicillin and its derivatives are manufactured worldwide.
VII. Conclusion: The Moral Imperative Not to Return to the Bad Old Days
“The future” has no pressure groups to represent it. The future has no lawyers to lobby for regulatory relief today. The lives saved in the future by today’s innovations have no voice. It is therefore vital that we learn from the COVID-19 outbreak to improve the future for our children and grandchildren by freeing up the healthcare sector to provide better service and more lifesaving breakthroughs.
Many of the healthcare shortages in California today, particularly labor shortages, could have been prevented had government regulations never been imposed. The COVID-19 surges would have been easier to manage had markets, not regulations, determined healthcare staffing over past decades.
Through their various liberalization decrees issued during the COVID-19 outbreak, government officials essentially admitted that current healthcare regulations harm patients. The stranglehold of regulations prevented the healthcare sector from responding quickly and efficiently to new circumstances during the initial months of the outbreak and beyond. The healthcare workforce artificially was constrained, PPE and other medical supplies were inadequate, and testing was botched from the start.
The rules protected incumbent manufacturers from new competition and protected healthcare facilities and practitioners from new entrants. Healthcare customers suffered as a result of that protectionism through higher prices, fewer providers and points of care, less innovation, lower quality of care, less access to care, and fewer lifesaving breakthroughs long term. Lives have been lost as a result.
Regulatory walls were built around the United States, around California, around providers and occupations, and even around tasks that skilled people perform—all to protect the politically connected from competition. Patient care suffered as a result.
Emergency liberalizations should be made permanent and the broader reforms discussed in the recommendations section should be adopted. If it was right to remove or relax healthcare regulations during the outbreak, it is right to eliminate those regulations forever. It would be immoral to act otherwise.
As William Sage, a professor of surgery (MD) and a professor of law (JD) at the University of Texas, Austin, said, “COVID has shown the difference between fact and assertion about our licensing restrictions and our geographical restrictions. ... There should be a presumption of permanence attached to all of these emergency liberalizations. And instead of the incumbents saying to the challengers ‘show us even more data than you have to support your position,’ now the challengers should say to the incumbents ‘show us data if you really want to revert to the bad old days.’”
If made permanent, liberalizations would make it easier to respond to the next healthcare crisis—and there will be another crisis someday. But those changes should be adopted permanently regardless of when the next crisis strikes. Reforms are the moral response because they will improve the lives of people today and of future generations by facilitating better access to affordable, high-quality care and more lifesaving breakthroughs, a win for patients regardless of the existence of a pandemic.
If it works during this crisis, it will work when the crisis is over.










